КЬЕЛЬДАЛЯ СПОСОБ
iP3
Рисунок 1.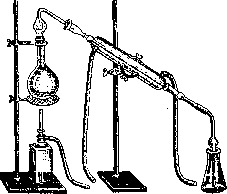
Рисунок 2.
ных аппаратов, где соединено несколько таких установок (рис. 3). Перегонная колба имеет объем в 600—800 см3. Она закрывается пробкой с предохранительной трубкой для улавливания щелочных брызг (рис. 4). Предварительным опытом с 5 см3 крепкой серной к-ты определяют, сколько 33%-ного едкого натра требуется для нейтрализации взятого для сжигания количества крепкой серной к-ты. Обычно для этого идет около 90 см3 раствора щелочи. Для подщелачива-ния перегоняемой жидкости берут нек-рый избыток (5—10 см3) щелочи. Содержимое колбы Кьельдаля осторожно разводят водой, переводят количественно в колбу для перегонки (общий объем жидкости—около 300 см3). Перегонку можно производить и прямо из колбы Кьельдаля, где производилось сжигание; в таком случае колба должна быть достаточно велика, а именно— объемом около 500 см3. При подщелачивании жидкости, предназначенной для перегонки, приливают не все нужное количество щелочи сразу, а только лишь % его> во избежание потери части аммиака из сильно разогревшейся жидкости; дают остыть. Тем временем в приемник с помощью пипетки отмеривают 50 см3 п/10 титрованной кислоты.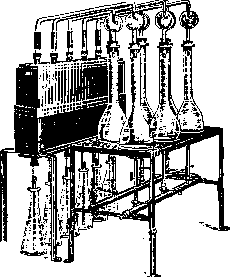
==£ЗУ
Рисунок 3. В к-ту погружают конец насадки, которой кончается холодильник. Когда жидкость в перегонной колбе остынет, в нее всыпают полную чайную ложку талька в порошке, что необходимо для равномерного кипения щелочной жидкости, вливают сразу оставшееся количество щелочи и тотчас же присоединяют колбу к заранее тщательно собранному перегонному аппарату. Жидкость в колбе взбалтывают, причем она меняет цвет, и в ней выделяется голубой осадок гидрата окиси меди Си(ОН)2, появление которого указывает на то, что прибавлено достаточное количество щелочи. Если при сжигании употреблялась ртуть, то кроме щелочи и талька в перегонную колбу следует добавить еще сернистой щелочи, K2S или Na2S, причем ртуть выпадает в виде сернистой ртути. Это необходимо для разрушения прочных соединений аммиака со ртутью. Удобнее всего с последней порцией щелочи влить 25 см3 5%-ного раствора K2S, отчего жидкость в колбе принимает черный цвет.—Теперь начинают осторожно нагревать жидкость и доводят до сильного кипения; жидкость должна кипеть ровно, без сильных толчков. При правильной перегонке к-ту должно засасывать лишь в добавочную трубку холодильника. Обыкновенно весь аммиак удается отогнать в продолжение 40—45 минут.
Рис.
Перед окончанием перегонки необходимо, чтобы дестилят обмыл внутренность трубки холодильника, в к-рую была засосана жидкость из приемника; для этого из-под приемника вынимают подкладки, на к-рых он стоял, так, чтобы конец холодильника оказался над к-той. Минут через 5, когда промоется холодильник, пробуют дестилят на полноту отгонки аммиака. Для этого на мгновение разобщают трубку холодильника и наконечника, смачивают красную лакмусовую бумажку каплей жидкости, стекающей по холодильнику; в случае хотя бы слабого посинения бумажки перегонку продолжают еще 15—20 минут. Титрование. Прибавив к полученной жидкости несколько капель индикатора (спиртового раствора метилрота 1:1.000 или настойки из 3 г кошенили в 250 см3 25%-ного спирта), производят титрование п/10 раствором едкого натра. Слепой опыт необходимо поставить для определения количества аммиака, содержащегося в реактивах. Обычно содержащийся в реактивах аммиак связывает лишь 0,1—0,5 ем3 п/10 кислоты. Вычисление. Предположим, на титрование в слепом опыте пошло 49,5 ом3 п/10 раствора NaOH, а при анализе—25,0 см3 того же раствора NaOH. Выделившийся аммиак связал следовательно количество к-ты, соответствующее (49,5-25,0) см* п/10 раствора NaOH. Т. к. 1 амзп/ю раствора NaOH соответствует 0,001401 г азота, то во взятом для анализа количестве вещества содержалось (49,5-25,0) 0,001401 г N. Полученный результат перечисляют на проценты. М и к р о к ь е л ь д а л ь по особенностям перегонки можно разделить на след. модификации. 1) Аммиак гонится с водой по типу макрокьельдаля из колбочки для сжигания. В этом случае особую опасность представляют толчки при кипении. 2) Аммиак гонится со струей пара, проходящего в Кьель-далевскую колбу из особого парообразователя (метод Бангаидр.). Для работы по этим методам требуется навык, иначе легко происходят неудачи от засасывания и перебрасывания содержимого колбы К. и приемника. 3) Аммиак отгоняется с помощью проса-сывания струи воздуха. Эти методы требуют довольно много времени для выполнения. 4) • Аммиак отгоняется при кипячении содержимого перегонной колбы и при просасы-вании в то же время струи воздуха (метод Pincussen'a); этот метод очень похож на нижеописываемый, в нем лишь сложнее соединение приемника с аппаратом. 5) Аммиак гонится с помощью пара в эвакуированной системе. Этот метод (Парнаса) предназначен для определения очень малых количеств аммиака, но в виду сложности аппаратуры едва ли удобен для широкого применения. Аммиак отгоняется при кипячении содержимого перегонной колбы и при пропускании струи воздуха, причем это достигается нагнетанием его. Этот метод представляет нек-рые преимущества и заключается в следующем. Сжигание субстрата производится в колбах К. емкостью в 100 см3 (иногда в колбах в 50 см3). К субстрату прибавляется 1 см3 крепкой H2S04 и несколько капель (до 8) 10%-ного раствора CuS04. Если же осаждение белков в анализируемой жидкости производилось реактивом Банга, то кроме H2S04 можно больше ничего не прибавлять, так как фосфорномолибденовая кислота сама по себе действует как катализатор. В последнее время Фойт (Voit) на том основании, что медь может задерживать в растворе аммиак, предложил пользоваться для сжигания пергидролем, добавляемым в количестве нескольких капель уже тогда, когда органические вещества почти сгорели и выделился уголь. Кроме этого для уменьшения толчков при пережигании но- лезно бросить в колбочку платиновую проволочку около 1 см длиной. Перегонка аммиака производится из аппарата, который очень похож на аппарат Банга (рисунок 5), но в котором вместо парообразователя находится газометр 1. Воздух из этого газометра поступает через промывалку с серной к-той в перегонную колбу совершенно так же, как пар из парообразователя в аппарате Банга. Кроме этого от макрокьель-даля аппарат отличается тем, что через воронку 2 можно впустить щелочь при закрытой колбе. Перед работой весь аппарат пропаривают. В остывшую колбу прибавляют 10 см3 воды и присоединяют ее к аппарату. Затем, если хотят вести определение иодометрически,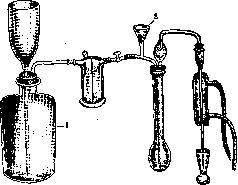
Рисунок 5.
берут раствор титрованной серной к-ты с прибавленным KJ03. Приготовляется этот раствор следующим образом: 10 см3 п/1в раствора HaS04 и 40 см3 нейтрального п/ю раствора KJO, отмеривают в мери-т ельную колбу емкостью в 100 см3 и доливают водой до метки. По отношению к серной к-те раствор— п/юо (центинормален). Такого раствора отмеряют в приемник-рюмку 2 см3 и погружают в него конец холодильника. В мерительный цилиндр наливаю^ 4—5 см3 насыщенного раствора NaOH, слитого с осад-' ка нерастворимой в нем соды и свободного следовательно от примеси ее. Это количество доводят до 10 см3 водой и полученный раствор осторожно впускают, открывая зажим, из воронки в перегонную колбу; непосредственно за тем открывают кран, ведущий к газометру, и в колбу пускают воздух с такой скоростью, чтобы пузырьки легко было считать. Под перегонную колбу подставляют горелку и жидкость доводят до сильного кипения, а через холодильник пускают сильный ток холодной воды. Благодаря пропускаемому из газометра воздуху жидкость кипит ровно, и к-та из приемника почти не засасывается в холодильник; в то же время пары аммиака вымываются из жидкости. По истечении 4 минут главная масса аммиака уже перегнана. Опуская рюмочку, вынимают конец холодильника из жидкости приемника и продолжают перегонку еще одну-две минуты, причем капли дестилята обмывают внутренность трубки. Кончая перегонку, следует попробовать капающий дестилят лакмусовой бумажкой, затем небольшим количеством воды из шприцфляша ополаскивают конец холодильника. Титрование. К дестиляту прибавляют 2 си3 5 %-ного раствора KJ, не содержащего свободного J, или прямо насыпают 0,1 г KJ в порошке, причем жидкость должна приобрести бурый цвет от выделившегося иода (если этого не происходит—аммиак нейтрализовал всю к-ту, опыт испорчен и надо в след. раз взять в приемник больше к-ты или более крепкую концентрацию ее или сжечь меньше вещества). Реакция выделения иода, идущая по уравнению 5KJ + + KJOa+6HCl=6KCl+3H20 + 6J, протекает не моментально; кроме того колич. выделившегося иода зависит несколько и от концентрации веществ. Поэтому, чтобы количество дестилята было всегда одинаково, важно перегонять определенное время, напр. 5—6 мин.; что же касается времени титрования, то рекомендуют его производить спустя 5 мин. после прибавления KJ. По истечении срока прибавляют 2—3 капли 1 %-ного раствора крахмала (1 г растворимого крахмала нагревают до растворения с 10—15 см3 воды, после чего раствор доливают до 100 см3 насыщенным раствором КС1; этот раствор может быть употребляем только до тех пор, пока он дает с очень разведенным раствором иода, чисто синюю окраску; если же окраска получается фиолетовая, то раствор не годен) и титруют п/200 рас- твором гипосульфита до исчезновения синего оттенка, не возвращающегося в течение первых минут. На посинение, наступающее спустя 10—15 минут, обращать внимание не следует. Раствор гипосульфита приготовляется непосредственно перед употреблением из п/ю раствора его. Одновременно производят слепой опыт при тех же условиях, какие имели место при анализе. Вычисление. Предположим, что при титровании в слепом опыте на 2 ом3 п/10в кислоты пошло 4,16 см 3 0,005121 п гипосульфита, а в основном опыте на то же количество к-ты пошло 1,95 см3 того же гипосульфита; следовательно перегнанный аммиак связал количество к-ты, соответствующее (4,16-1.95) см3 0,005121 п гипосульфита, или (4,16-1,95) 0,005121 см3 нормального раствора гипосульфита. Так как 1 см3 нормального раствора гипосульфита при этом титровании соответствует 0,01401 г, или 14,01 мг N, то найденное количество см3 нормального раствора гипосульфита соответствует (4,16-1,95).0,005121.14,01 мя N. Обычно количество азота выражают в мг%. Если хотят вести определение аммиака не иодометрически, а ацидометрически, то в таком случае в приемник-рюмку отмеривают 2—3 cmz п/100 НО и для титрования бе-РУТ п/юо раствор NaOH, свободный от соды, и раствор метилрота в 90-процентном спирте (1:10.000). Что касается полумикро метода, то он особенно применим при определении общего количества N в крови и моче; крови берется 0,1 см3 или 1 см3, разведенной в 25 раз; самое определение может быть произведено в аппарате, предназначенном для микроопределения, но только при этом следует брать больше кислоты для сжигания, больше щелочи для подщелачивания, употреблять п/50 кислоту в приемнике., дольше гнать, титровать ацидометрически и раствором не слабее n/so, Лит.: kj el da hi J., Neue Methode zur Be-stimmung des Stickstoffs In organischen Korpern, Ztschr. i. analytiscne Chemie, B. XXII, 1883; Gunning J., tlber eine Modification der Kjeldahl-Me-thode, ibid., B. XXVIII, 1889. Г. Дервиз. Смотрите также:
Смотрите также:
- КЭРТЕ Вернер (Werner Korte, родился в 1853 году), видный нем. хирург. Почти вся деятельность К. прошла вне академической работы в б-цах Берлина, где он своими блестящими работами и широкой эрудицией скоро ...
- КЮВЬЕ Жорж (Georges Cuvier, 1769— 1832), знаменитый сравнительный анатом и палеонтолог. Родился в Монбеляре (Эльзас). В 1788 г., будучи домашним учителем, начал занятия по естествознанию и изучал морск. животных, особенно моллюсков ...
- КЮММЕЛЬ Герман (Hermann Kummell, род. в 1852 г.), известный германский хирург, ученик Шеде. К. большую часть своей деятельности посвятил работе в б-цах города Гамбурга и создал целую плеяду учеников, имеющих уже ...
- КЮРИ Мария и Пьер. Кюри Мария, урожденная Склодовская (родилась в 1867 году). После окончания курса гимназии в Варшаве продолжала свои работы в Париже на физико-математическом факультете. В1895г., после выхода замуж за ...
- КЮТНЕР Герман (Hermann Kiittner, род. в 1870 году), хирург, профессор Бреславль-ского ун-тета (Германия), ученик Брунса. Автор многочисленных трудов из разных областей хирургии: ряд работ по военно-полевой хирургии относится преимущественно к началу ...